

News
-
2026 April: Xu-Kai, Yi-Lin and Li have contributed an article in Nature. Congratulations!
-
2026 February: Yao-Qi, Fang and Li have contributed an article in Cell Research. Congratulations!
-
2026 January: Li has been selected as one of the 2025 Most Cited Chinese Researchers (released by Elsevier). Congratulations to Li!
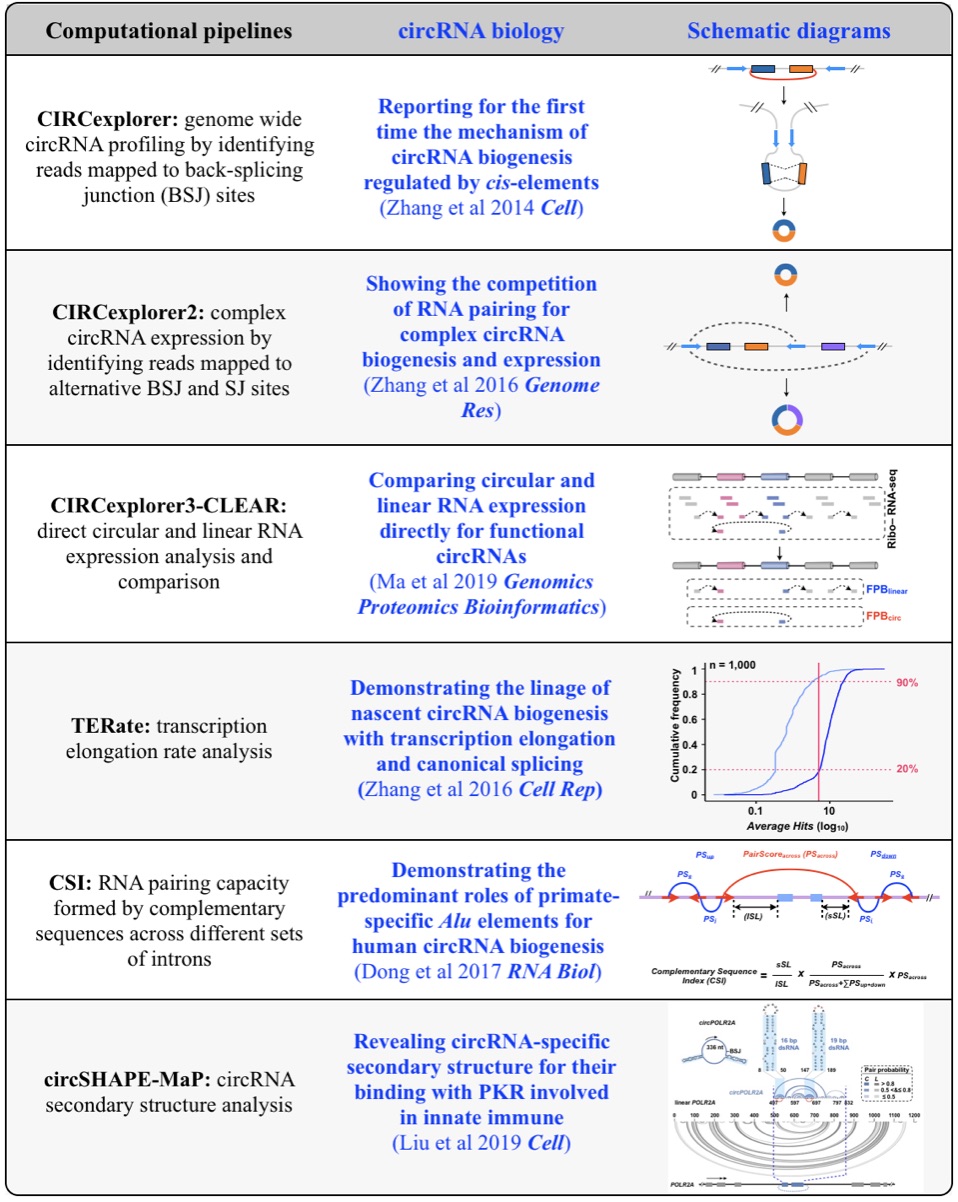
Fig 1. Computational pipelines and biology of circRNAs.
1. Genome-wide profiling and characterization of functional circRNAs
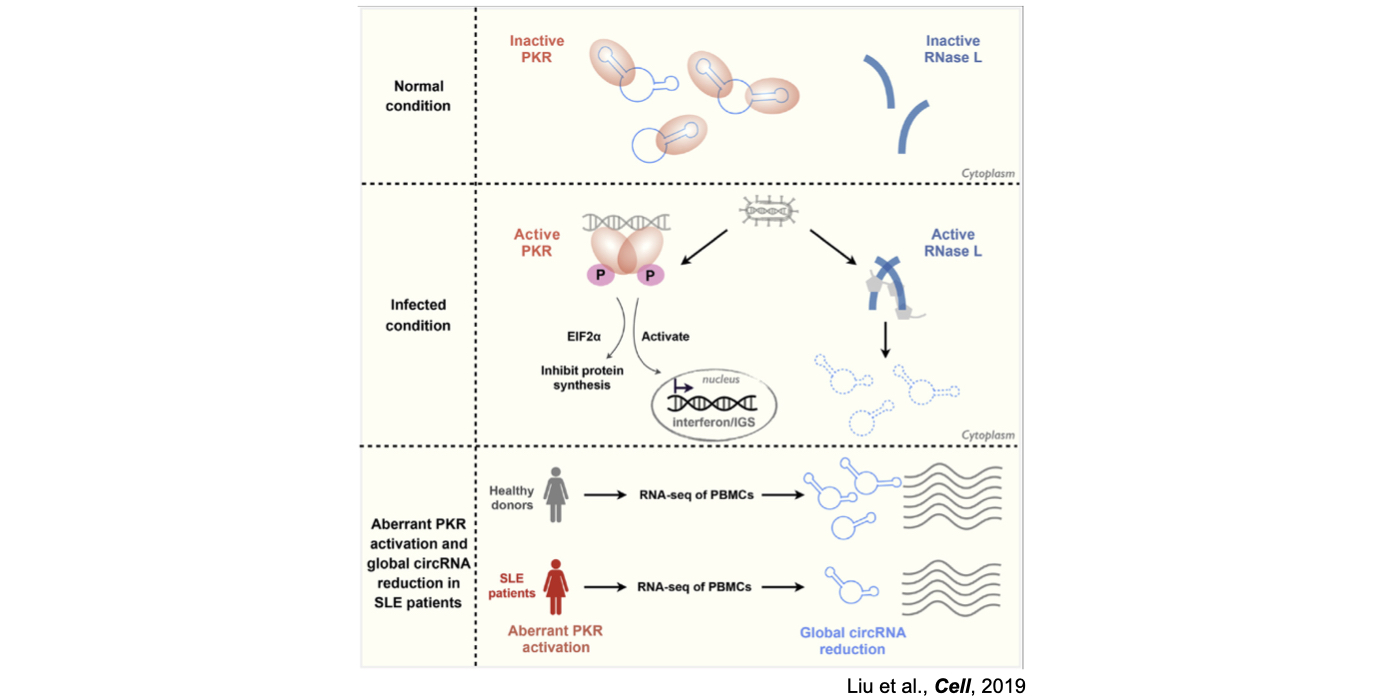
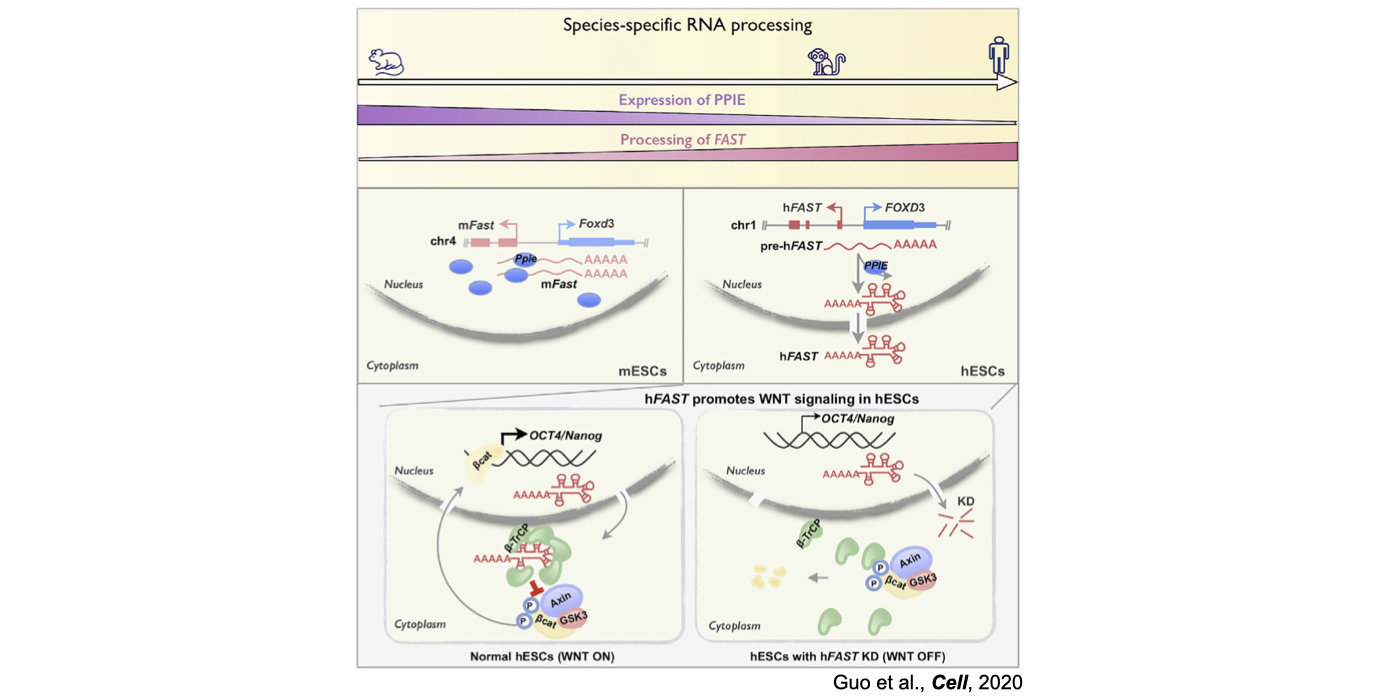
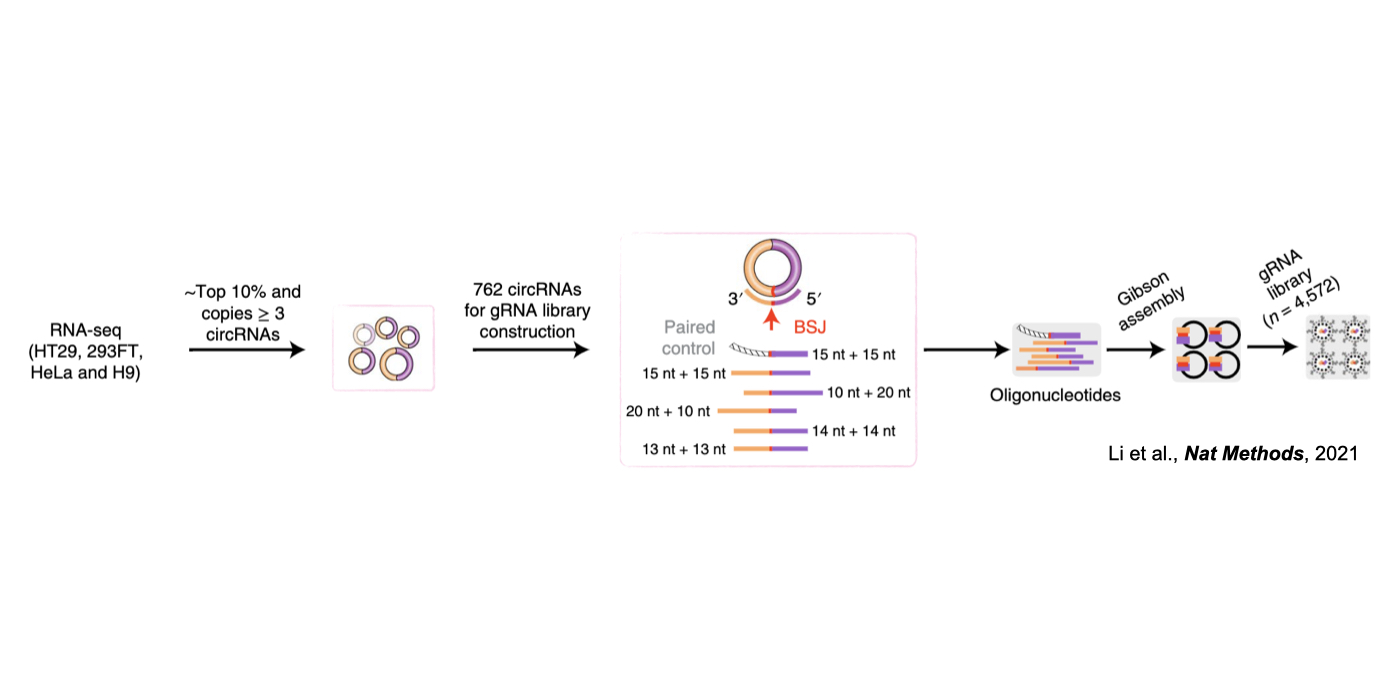
Although only about 2% of the human genome encode protein sequences, recent advances in genome-wide analyses have revealed that the majority of the human genome are transcribed, largely from noncoding segments that were used to be considered as “junk sequences” or “dark matter” of the human genome. Besides well-characterized housekeeping noncoding RNAs (ncRNAs, such as tRNA, rRNA, snRNA and snoRNA) and small regulatory ncRNAs, the transcriptome has become even more complex with pervasively transcribed lncRNAs (at least 200 nt long without coding potential). By collaborating with experimental biologists, we have pioneered in developing poly(A)– RNA-seq and revealed a series of novel types of lncRNAs with specific structural features, including snoRNA-related lncRNAs (sno-lncRNAs; Yin et al 2012 Mol Cell; Zhang et al 2014 BMC Genomics), circular intronic RNAs (ciRNAs; Zhang et al 2013 Mol Cell), 5’ snoRNA-capped and 3’-polyadenylated lncRNAs (SPAs; Wu et al 2016 Mol Cell) and covalently-closed circRNAs produced from back-splicing of exons (circRNAs; Zhang et al 2014 Cell). Here, I mainly summarize systematic findings and our current understanding of circRNA biogenesis and functions, identified in my lab with collaboration (Fig.1).
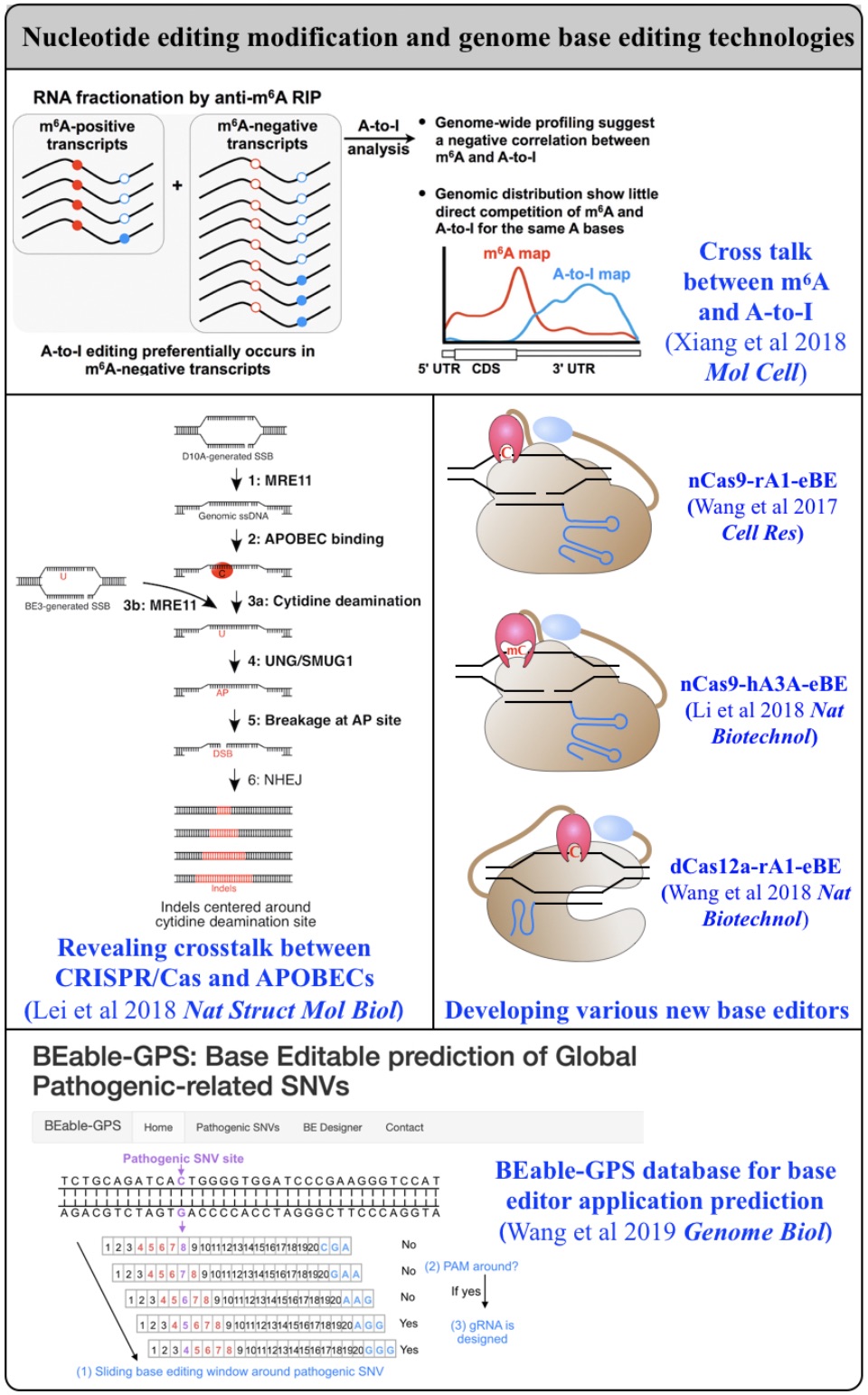
Fig 2. From editing biology to editing technology.
2. Nucleotide editing modification and genome base editing technologies
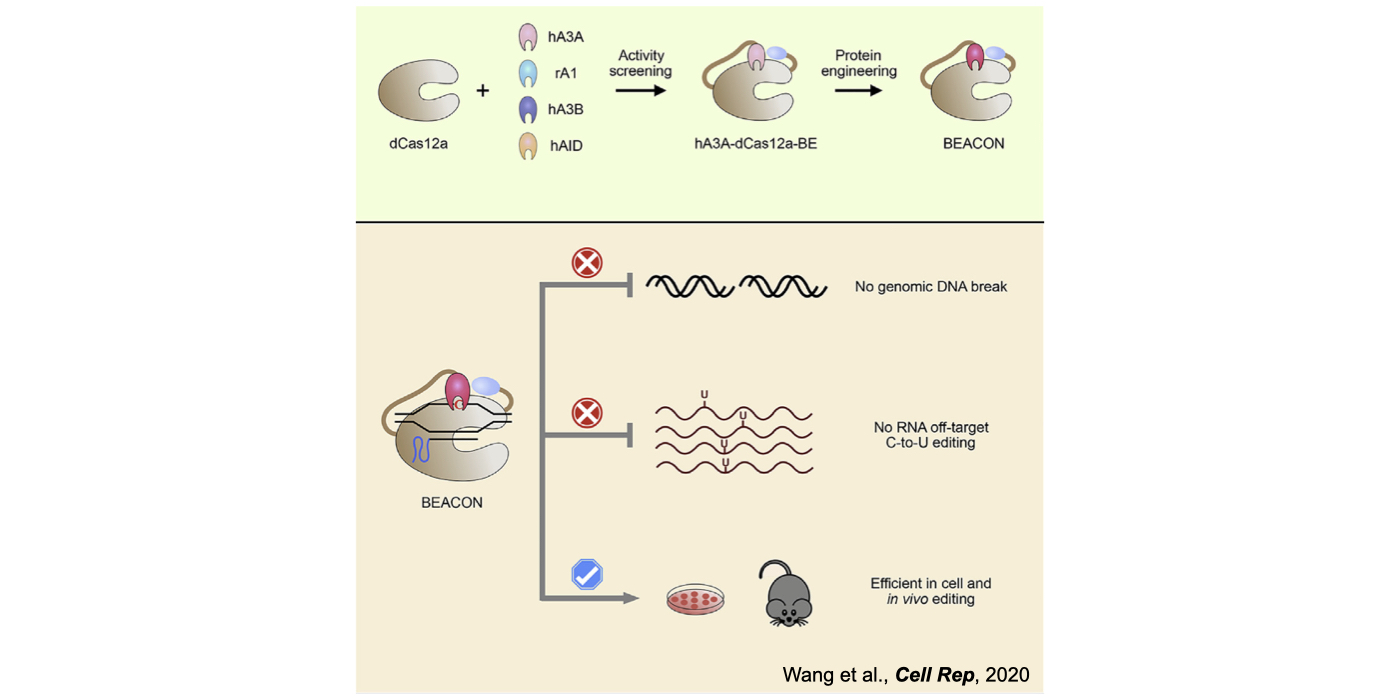
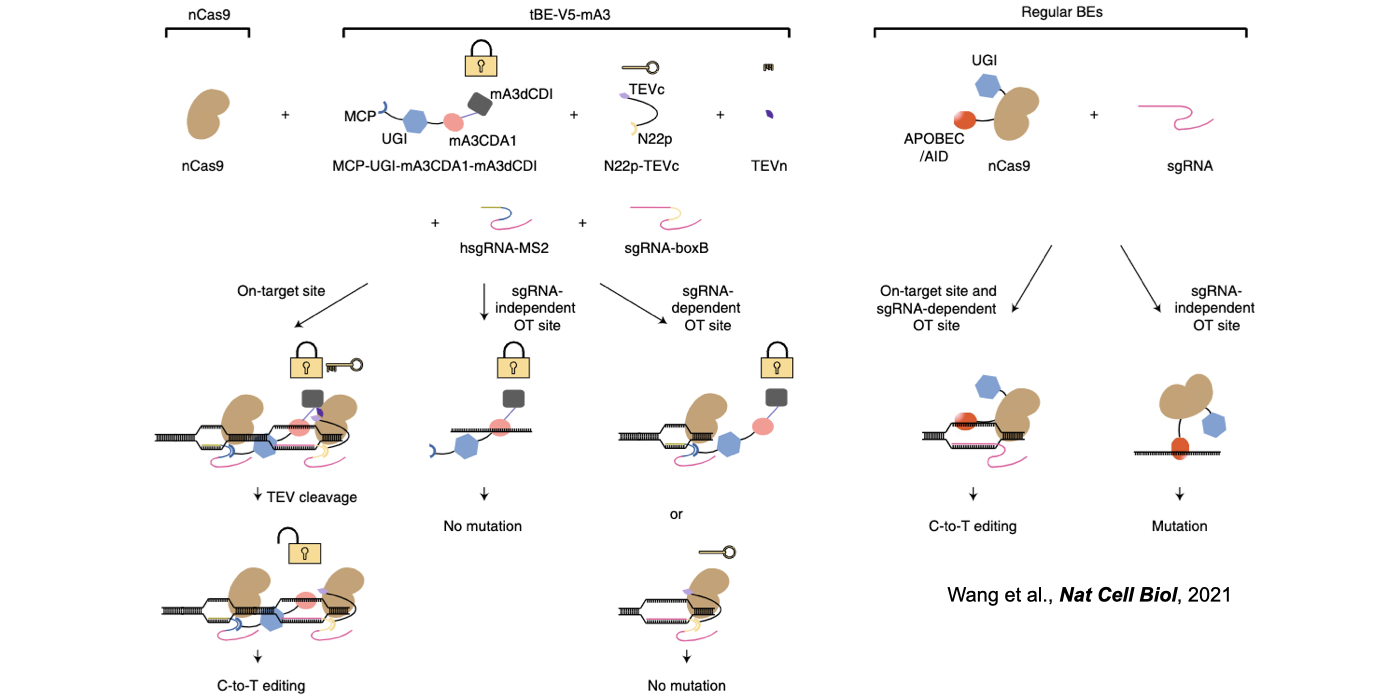
Biochemical editing/modification on DNA/RNA not only expands the number of functions encoded by our genomes but also provides additional mechanisms of gene regulation. Over 100 different types of RNA editing/modifications have been identified transcriptome-wide. The most predominant form of such editing/modifications in higher eukaryotes is adenosine-to-inosine (A-to-I) editing on RNA, catalyzed by members of adenosine deaminases acting on RNA (ADAR) enzyme family. As the resulting I preferentially bases pair with cytidine (C) and is therefore functionally guanosine (G), A-to-I editing can have profound effects on downstream RNA processing and function, including recoding of open reading frames, altering the pattern of alternative splicing, interfering with microRNA function, modulating RNAi activity, and playing other roles in gene regulation. In addition to adenosine deaminases, cytidine deaminases are also known to be responsible for cytidine deamination on both single strand DNA (ssDNA) and RNA, catalyze by the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like/activation-induced cytidine deaminase (APOBEC/AID) family of enzymes. Another major focus of my lab is developing new computational pipelines to globally profile nucleobase editing/modification events and to inspect potential crosstalk between different types of editing/modifications (Xiang et al 2018 Mol Cell). Importantly, by underscoring a previously underappreciated linkage between APOBECs with clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR associated system (Cas) 9-generated DNA breaks, my lab has contributed in the genome editing field by generating a variety of base editing systems to perform C-to-T changes at target genomic sites in broad genome backgrounds (Lei et al 2018 Nat Struct Mol Biol; Li et al 2018 Nat Biotechnol; Wang et al 2018 Nat Biotechnol; Wang et al 2019 Genome Biol; Wang et al 2020 Cell Rep) (Fig. 2).
Research Summary
The completion of the human genome project (HGP) in the beginning of this century and the application of affordable high-throughput sequencing technologies in the past decade have led life science researches to the post-genome era. The resulting wealth of deep-sequencing datasets at genome (including epi-genome) and transcriptome (including epi-transcriptome) levels challenges us to fully understand how functional genomic elements are transcribed and regulated, thus leading to human health and/or diseases. Importantly, the advent of novel genome editing technologies provides us powerful methods to change genetic information at desired target sites at single nucleotide resolution, which benefits not only basic research aiming to decipher how different genotypes result in distinct phenotypes but also pre-clinical study potential to cure human diseases caused by genetic mutations.
Since established in 2011, my lab has continuously developed and applied computational strategies, including the-state-of-the-art machine learning and deep learning approaches, together with deep-sequencing technologies and diverse biochemical methods,
- to dissect complex expression, novel function and potential application of long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) from back-splicing;
- to achieve precise genetic changes in the application of biomedical research and therapeutics by the invention of novel genome base editing systems;
- to uncover previously-underappreciated modes of RNA regulation, such as alternative polyadenylation and subcellular localization, at single-cell resolution.
Research Achievements
1. Genome-wide profiling and characterization of functional circRNAs
2. Nucleotide editing modification and genome base editing technologies
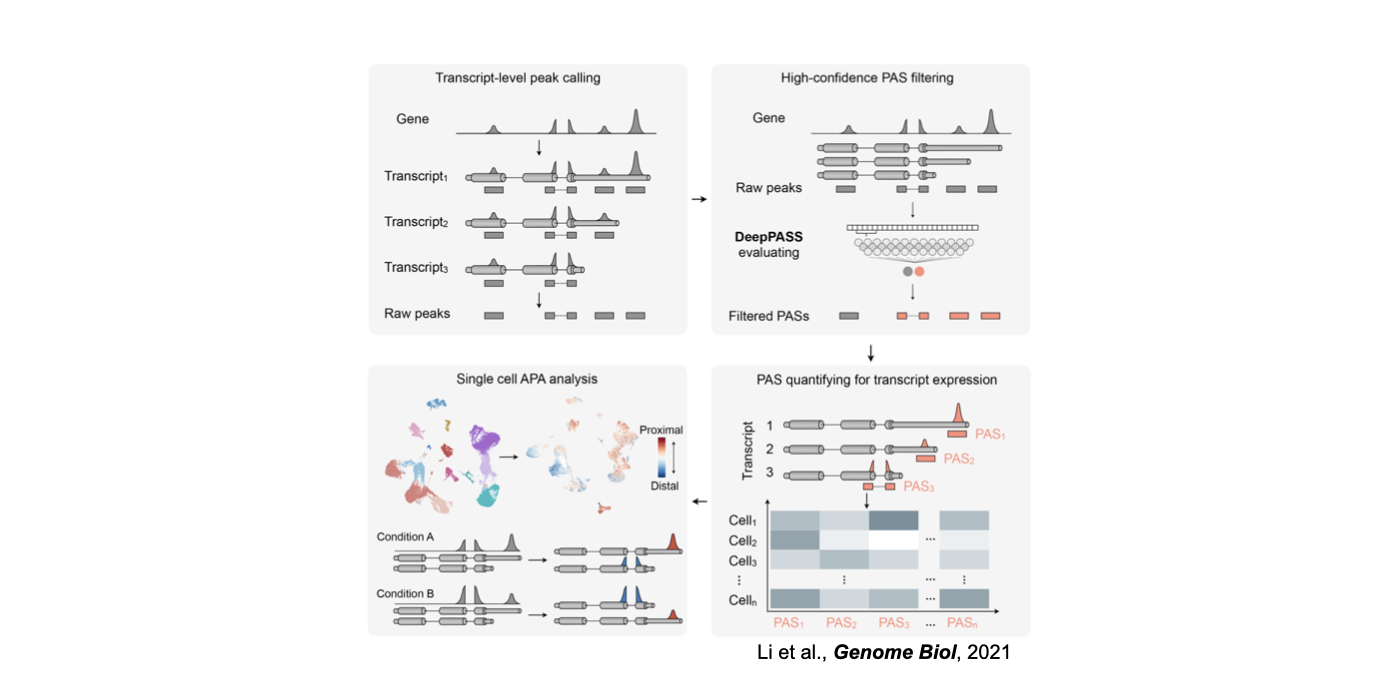
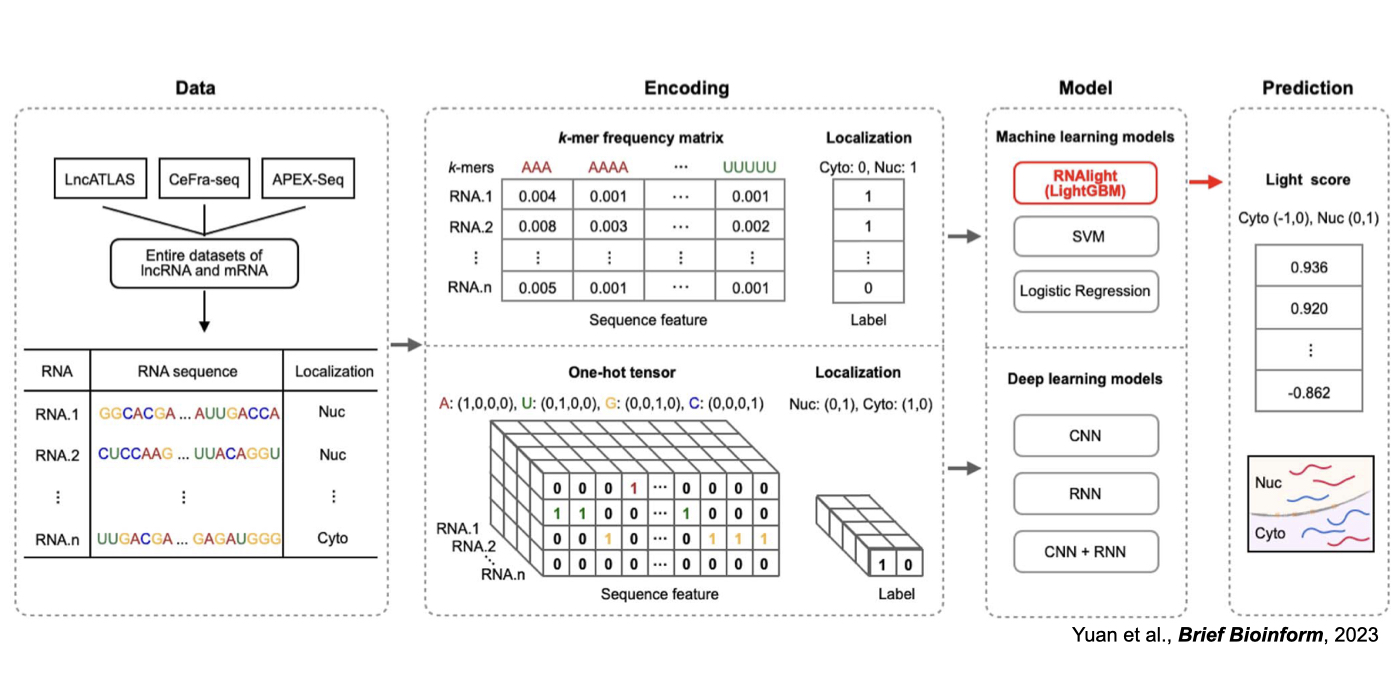
3. Novel machine/deep learning approaches for the understanding of RNA regulation
Collaborators
Prof. Dr. Ling-Ling Chen, SIBCB, SIBS, CAS
Contact
Address: 科研二号楼CB2-044-046, 131 Dong An Road, Shanghai 200031, P.R.China
Telephone: (+86)-021-54661081
Email: liyang_fudan@fudan.edu.cn